Alexander disease
Epidemiology
Epidemiology
Alexander disease is a rare, genetic, progressive astrocytopathy that often presents as a leukodystrophy in children and as a neurodegenerative disease in adults.1,20 The prevalence has been estimated at 1 case per 1 to 3 million people based on limited evidence3,21,22; however, it may be underreported due in part to people with Alexander disease being misdiagnosed with other more prevalent diseases.3,4
Alexander disease impacts people of all ages,2,6-8 with approximately two-thirds of cases observed in infants and juveniles and one-third in adults.23 There is no racial, ethnic, geographic, or sex predominance, as it is primarily caused by de novo pathogenic variants of glial fibrillary acidic protein (GFAP). Pathogenic variants of GFAP exhibit an autosomal dominant pattern of inheritance. While the majority of cases result from de novo mutations, familial cases are increasingly being recognized as diagnostic capabilities improve.24

Alexander disease accounts for approximately

of leukodystrophies—a group of rare genetic disorders that affects white matter in the central nervous system.22,25-29
Etiology
An astrocytopathy, Alexander disease predominantly arises from de novo pathogenic variants in GFAP that disrupt the structure and function of astrocytes in the central nervous system, resulting in abnormal myelination and neuronal dysfunction.1,5,20,30,31
Symptoms
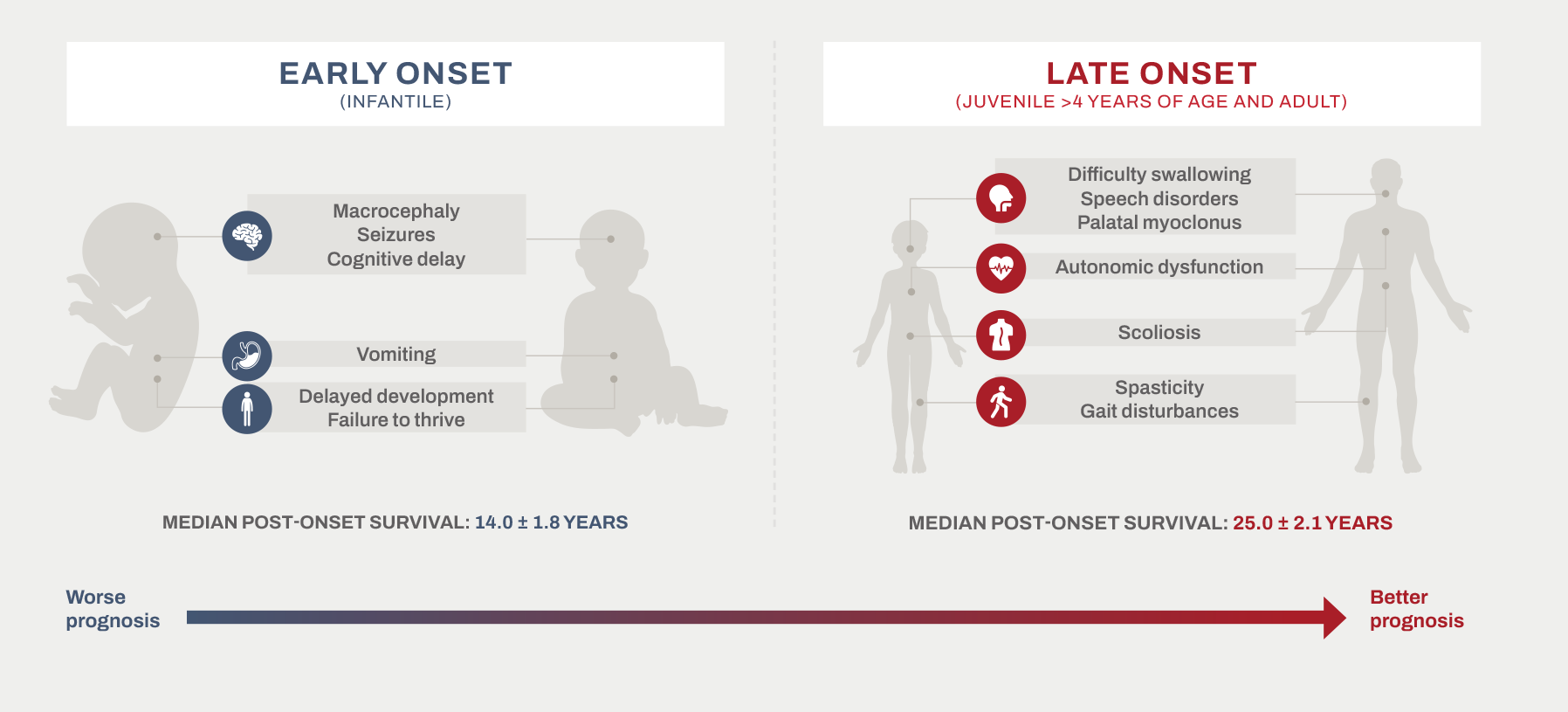
Alexander disease is a heterogeneous disease that presents differently depending on the age of onset2-4,32